The proteomes of 100 organisms across the tree of life
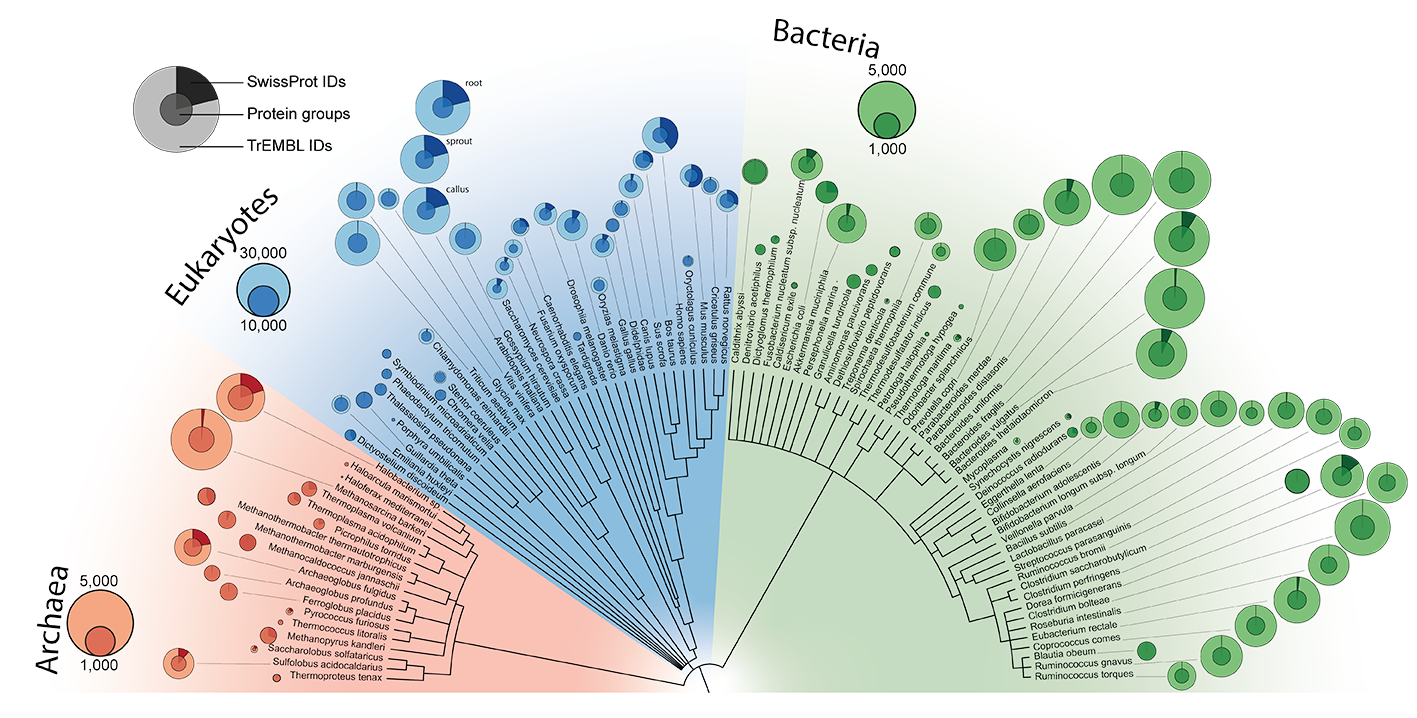
Proteins perform the vast majority of functions in all biological domains but their large-scale investigation has lagged behind due to technological challenges. Since the first essentially complete eukaryotic proteome had been reported, advances in mass spectrometry (MS)-based proteomics have enabled increasingly comprehensive identification and quantification of the human proteome across a wide variety of experiments. However, apart from human samples only a limited number of model organisms has been investigated by proteomics and there are few comparisons across species, especially compared to genomics initiatives.
In this work, we employ an advanced proteomics workflow, in which the peptide separation step is performed by a microstructured and extremely reproducible chromatographic system for the in-depth measurement of 100 taxonomically distinct organisms. With two million peptides and 340,000 stringent protein identifications obtained in a standardized manner, we doubled the number of proteins with solid experimental evidence known to the scientific community. The data provide an important and large-scale use case for sequence-based machine learning, as we demonstrate by experimentally confirming predicted peptide properties of Bacteroides uniformis.
The extensive acquired dataset provide the research community with a comparative view into the functional organization of organisms across the entire evolutionary range, which can be explored and functionally compared with this webpage.